ΤΗΣ ΑΝΘΗΣ ΔΡΟΥΣΙΩΤΟΥ*
Η κλασσική γαλακτοζαιμία είναι ένα κληρονομικό μεταβολικό νόσημα που επηρεάζει το μεταβολισμό της γαλακτόζης, του βασικού μονοσακχαρίτη στο γάλα και τα γαλακτοκομικά προϊόντα. Η ασθένεια οφείλεται στην ανεπάρκεια του ενζύμου ουριδυλο-τρανσφεράση της 1-φωσφορικής γαλακτόζης, λόγω μεταλλάξεων στο αντίστοιχο γονίδιο GALT, και κληρονομείται με αυτοσωμικό υπολειπόμενο τρόπο. Οι κλινικές εκδηλώσεις στην κλασσική γαλακτοζαιμία οφείλονται στην συσσώρευση της γαλακτόζης και των μεταβολιτών της στους ιστούς, σαν αποτέλεσμα της ενζυμικής ανεπάρκειας. Τα νεογνά είναι φυσιολογικά κατά τη γέννηση και τα συμπτώματα εμφανίζονται με την έναρξη της σίτισης με γάλα, και περιλαμβάνουν ηπατική νόσο, ίκτερο, στασιμότητα βάρους, γαστρεντερικά προβλήματα, λήθαργο και υπογλυκαιμία. Τα βρέφη με κλασσική γαλακτοζαιμία συχνά παρουσιάσουν και καταρράκτη. Αν δε διαγνωσθεί έγκαιρα η ασθένεια οδηγεί σε σοβαρά ηπατικά, νεφρολογικά και νευρολογικά προβλήματα και σε πρόωρο θάνατο. Η αντιμετώπιση συνίσταται στην αφαίρεση της γαλακτόζης από τη διατροφή, κάτι που πρέπει να συνεχισθεί εφόρου ζωής. Η αποτελεσματικότητα της δίαιτας ελέγχεται με την μέτρηση της 1-φωσφορικής γαλακτόζης στα ερυθρά αιμοσφαίρια.
Στη συγκεκριμένη μελέτη προσδιορίστηκε η συχνότητα των φορέων της κλασσικής γαλακτοζαιμίας στον Ελληνοκυπριακό πληθυσμό και πραγματοποιήθηκε ο μοριακός χαρακτηρισμός των παθολογικών αλληλόμορφων. Η επιδημιολογική μελέτη πραγματοποιήθηκε σε δείγμα 528 Ελληνοκυπρίων με καταγωγή από όλες τις επαρχίες της Κύπρου. Οι φορείς εντοπίστηκαν μέσω του προσδιορισμού της δραστικότητας του ενζύμου GALT στα ερυθρά αιμοσφαίρια και ακολούθως διερευνήθηκαν μοριακά.
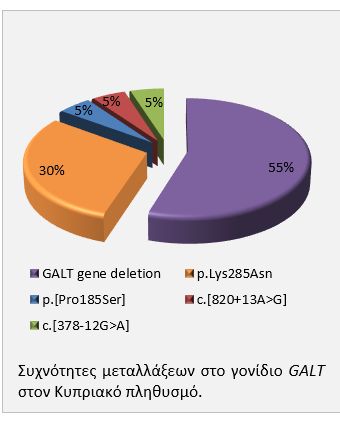
Εντοπίστηκαν συνολικά 5 μεταλλάξεις σε ασθενείς και φορείς της κλασσικής γαλακτοζαιμίας: απάλειψη 8,5kb, η οποία περιγράφηκε προηγουμένως από το εργαστήριο μας, σε ποσοστό 55% των αλληλομόρφων, οι γνωστές μεταλλάξεις p.Lys285Asn (30%), p.Pro185Ser (5%), και c.820+13A>G (5%) καθώς και η νέα μετάλλαξη c.378-12G>A (5%). Η απάλειψη 8,5kb είναι μια καινούργια μετάλλαξη που δεν έχει βρεθεί σε άλλους πληθυσμούς και παρουσιάζει ιδιαίτερο ενδιαφέρον γιατί επηρεάζει όχι μόνο το γονίδιο της γαλακτοζαιμίας αλλά και το επόμενο γονίδιο, αυτό του υποδοχέα α της ιντερλευκίνης 11 (IL11RA). Μεταλλάξεις στο γονίδιο IL11RA προκαλούν κρανιοσυνώστοση. Οι Κύπριοι ασθενείς με γαλακτοζαιμία οι οποίοι είναι ομόζυγοι για αυτή την απάλειψη έχουν ταυτόχρονα και κρανιοσυνώστοση. Ενδιαφέρον επίσης αποτελεί το γεγονός ότι η μετάλλαξη p.Gln188Arg, η οποία είναι η συχνότερη σε Ευρωπαϊκούς πληθυσμούς, δεν ανιχνεύθηκε στο συγκεκριμένο δείγμα του Κυπριακού πληθυσμού.
Η συχνότητα των φορέων της κλασσικής γαλακτοζαιμίας στον Ελληνοκυπριακό πληθυσμό προσδιορίστηκε σε 1:88 και η επίπτωση της νόσου εκτιμήθηκε σε 1 στις 31 000 γεννήσεις, ψηλότερη από την μέση παγκόσμια επίπτωση που είναι 1:62 000. Οι παραλλαγές στο γονίδιο της GALT οι οποίες μπορεί να παρουσιάζουν ηπιότερη συμπτωματολογία, και είναι γνωστές ως Duarte 1 και Duarte 2, ανιχνεύθηκαν σε ποσοστό 5.5% και 2.5%, αντίστοιχα.
Τα αποτελέσματα της μελέτης αυτής θα μας βοηθήσουν να βελτιώσουμε τις υπηρεσίες μας προς τους Κύπριους ασθενείς με γαλακτοζαιμία και τις οικογένειες τους.
Το πρόγραμμα χρηματοδοτήθηκε από το Ίδρυμα Προώθησης Έρευνας (ΠΕΝΕΚ 06/09/64) και το Telethon Cyprus.
*Διευθύντρια Τμήματος Βιοχημικής Γενετικής
Καθηγήτρια Σχολής Μοριακής Ιατρικής Κύπρου
Δημοσιεύσεις:
- “Classic galactosaemia in the Greek Cypriot population: An epidemiological and molecular study”. R. Papachristoforou, P. Petrou, H. Sawyer, M. Williams, A. Drousiotou. Ann Hum Genet. 2019;1-8. doi: 10.1111/ahg.12318.
- "A Novel Large Deletion Encompassing the Whole of the Galactose-1-Phosphate Uridyltransferase (GALT) Gene and Extending into the Adjacent Interleukin 11 Receptor Alpha (IL11RA) Gene Causes Classic Galactosemia Associated with Additional Phenotypic Abnormalities". R. Papachristoforou, P. P. Petrou, H. Sawyer, M. Williams, A. Drousiotou (2014) JIMD Rep. 2014;12:91-8. doi: 10.1007/8904_2013_249.