H Γενετική του Συνδρόμου Πολλαπλής Ενδοκρινούς Νεοπλασίας τύπου 2 (MEN2) στην Κύπρο και το φαινόμενο της Αρχής του Ιδρυτή
Το σύνδρομο της Πολλαπλής Ενδοκρινούς Νεοπλασίας τύπου 2 (ΜΕΝ2) κληρονομείται με τον αυτοσωματικό επικρατούντα χαρακτήρα και οφείλεται σε μεταλλάξεις στο πρωτο-ογκογονίδιο RET στο χρωμόσωμα 10. Το πρωτο-ογκογονίδιο RET κωδικοποιεί τον διαμεμβρανικό υποδοχέα της κινάσης της τυροσίνης και οι μεταλλάξεις οδηγούν σε νόσους των παραθυρεοειδών και του μυελού των επινεφριδίων.
Το σύνδρομο περιλαμβάνει τις υποκατηγορίες ΜΕΝ2Α, ΜΕΝ2Β και το οικογενές μυελώδες καρκίνωμα του θυρεοειδούς (FMTC). Στην υποκατηγορία ΜΕΝ2Α, το μυελοειδές καρκίνωμα (MTC) παρατηρείται στο 50% των ασθενών, το φαιοχρωμοκύττωμα σε ποσοστό 25-50% και υπερπαραθυρεοειδισμός σε ποσοστό 20-25%. Ασθενείς της ΜΕΝ2Β εμφανίζουν ΜΤC και φαιοχρωμοκύττωμα, αλλά όχι υπερπαραθυρεοειδισμό. Επιπλέον εμφανίζουν νευρινώματα βλεννογόνων και εκδηλώσεις συνδρόμου Marfan.
Η ταυτοποίηση μετάλλαξης στο πρωτο-ογκογονίδιο RET υποδηλώνει ότι το υπό εξέταση άτομο θα εμφανίσει MTC με πιθανότητα μεγαλύτερη του 90%.
Το τμήμα Μοριακής Γενετικής Λειτουργίας και Θεραπείας (ΜΓΛΘ) του Ινστιτούτου Νευρολογίας και Γενετικής (ΙΝΓΚ) ήταν ο συντονιστής του ερευνητικού έργου και έλαβαν μέρος ο διευθυντής του τμήματος καθηγητής Λεωνίδας Α Φυλακτού, ο δρ Παύλος Φάνης και ο δρ Βάσος Νεοκλέους. Επίσης συνεργάστηκαν από το ΙΝΓΚ ο κλινικός γενετιστής δρ Γιώργος Ταντελές, η κα Έλενα Σπανού-Αριστείδου, ο καθηγητής γενετιστής Μάριος Καριόλου, η δρ Μαρίνα Κλεάνθους και οι γενετιστές Γιώργος Χριστόπουλος και Παναγιώτης Μανώλη. Από την παιδιατρική κλινική του Μακάριου Νοσοκομείου συνεργάστηκαν η δρ Βιολέτα Αναστασιάδη και δρ Στέλλα Νικολάου. Από το ογκολογικό κέντρο της Τράπεζας Κύπρου ο πυρηνικός ιατρός δρ Σάββας Φράγκος και από την Ιατρική Σχολή του St George του πανεπιστημίου Λευκωσίας ο καθηγητής παιδοενδοκρινολόγος Νίκος Σκορδής. Τέλος συνέβαλαν επίσης η ενδοκρινολόγος δρ Έλενα Ανδρέου και ο μεταπτυχιακός φοιτητής Μιχάλης Μαυρομμάτης.
Η παρούσα μελέτη περιγράφει Κύπριους ασθενείς με κλινικά χαρακτηριστικά ΜΕΝ2 που υποβλήθηκαν σε γενετικό έλεγχο για το πρωτο-ογκογονίδιο RET μεταξύ των ετών 2002 και 2017.
Για τους σκοπούς της μελέτης σαράντα ασθενείς υποβλήθηκαν σε γενετική ανάλυση της αλληλουχίας των βάσεων του πρωτο-ογκογονιδίου RET. Οι ασθενείς, 23 γυναίκες και 17 άνδρες, προέρχονταν από 11 μη σχετιζόμενες κυπριακές οικογένειες όπως επίσης και από δύο σποραδικές περιπτώσεις. Η πλειοψηφία των Κυπρίων ασθενών σε ποσοστό 69,2% ήταν φορείς της μετάλλαξης p.Cys618Arg,ακολουθούμενη σε ποσοστό 7,7% από τις μεταλλάξεις p.Cys634Phe, και delE632-L633. Σε ποσοστό 15.4% εντοπίστηκε και η γνωστήp.Met918Thr που ως επί το πλείστο είναι υπεύθυνη για τον κλινικό φαινότυπο της υποκατηγορίας ΜΕΝ2Β. Η μέση ηλικία των ασθενών κατά τη διάγνωση του MTC που έφεραν την μετάλλαξη p.Cys618Arg ήταν 36,8 ± 14,2 έτη. Η ηλικία της διάγνωσης για το φαιοχρωμοκύττωμα κυμάνθηκε από τα 26 έως τα 43 έτη και σε 5 από τις 36 (13,9%) περιπτώσεις εμφανίστηκε ταυτόχρονα με το MTC.
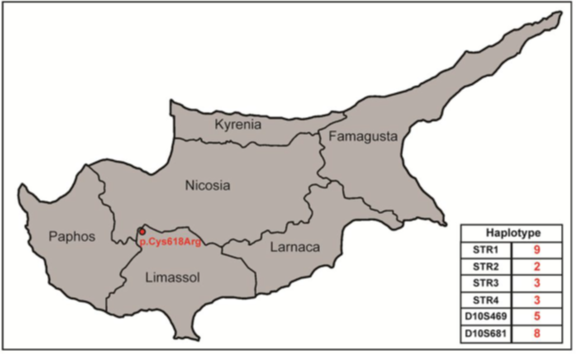
Ο γενετικός έλεγχος στα πλείστα μέλη των οικογενειών από τις εννέα οικογένειες που φέρουν την μετάλλαξη p.Cys618Arg έδειξε την παρουσία του ίδιου γενετικού ελαττώματος σε τριάντα έξι άτομα όλων των ηλικιών, εκ των οποίων 24 υποβλήθηκαν σε προφυλακτική ολική θυρεοειδεκτομή. Το οικογενειακό ιστορικό σε πέντε από τις εννέα οικογένειες με την μετάλλαξη p.Cys618Arg αποκάλυψε κοινή καταγωγή από χωριό στο βορειοδυτικό άκρο της επαρχίας Λεμεσού στην Κύπρο (Εικόνα 1).
Η υψηλή συχνότητα της μετάλλαξης p.Cys618Arg καθώς και οι κοινή προγονική καταγωγή των περισσότερων οικογενειών που τη φέρουν υποδηλώνουν πιθανό ιδρυτικό προγονικό συμβάν (founder effect phenomenon). Για τη διερεύνηση του ιδρυτικού φαινομένου της μετάλλαξης p.Cys618Arg διενεργήθηκε περαιτέρω διερεύνηση με τη χρήση γενετικών δεικτών (STRs). Ανάλυση του απλότυπου με τη χρήση STRs που διεξήχθη σε οικογένειες με ή χωρίς την μετάλλαξη p.Cys618Arg αποκάλυψαν έναν κοινό απλότυποσε όλους τους ασθενείς που έφεραν τη συγκεκριμένη μετάλλαξη.
Το φαινόμενο του ιδρυτή (founder effect) που καταδεικνύεται μέσα από τα αποτελέσματα της παρούσας μελέτης είναι συχνό φαινόμενο που παρατηρείται στον σχετικά ομοιογενή πληθυσμό της Κύπρου. Παρόμοια ιδρυτικά φαινόμενα μεταλλάξεων έχουν παρατηρηθεί και σε μελέτες που επίσης διεξήγαγε το τμήμα ΜΓΛΘ για άλλες ενδοκρινολογικές κληρονομικές παθήσεις όπως η ασθένεια της ανεπάρκειας της 5α-αναγωγάσης τύπου 2 (SRD5A2 gene) 1 και η Συγγενής Υπερπλασία των Επινεφριδίων 2 . Λόγω των σημαντικών συσχετίσεων που έχουν καταδειχτεί μεταξύ των διαφόρων μορφών ΜΕΝ2 (ιδιαίτερα της επιθετικότητας του MTC) με συγκεκριμένες μεταλλάξεις στο γονίδιο RET, η ταυτοποίηση των μεταλλάξεων είναι εξαιρετικά σημαντική για την επιβεβαίωση της διάγνωσης και τη θεραπευτική αγωγή που θα ακολουθηθεί.
Η ανάλυση αλληλουχίας των βάσεων του γονιδίου RET αποτελεί τη μοναδική απόλυτα έγκυρη εργαστηριακή ταυτοποίηση του συνδρόμου ΜΕΝ2. Ιδιαίτερα στην περίπτωση παιδιών με οικογενειακό ιστορικό, που δεν έχουν ακόμη βιοχημικά ή κλινικά ευρήματα για ΜΤC, η ανίχνευση και ταυτοποίηση συγκεκριμένων μεταλλάξεων στο πρωτο-ογκογονιδίο RET είναι καθοριστική για την επιλογή της ηλικίας για προφυλακτική ολική θυρεοειδεκτομή. Με αυτόν τον τρόπο η μείωση του κινδύνου που επιτυγχάνεται είναι εξαιρετικά μεγάλη.
Εικόνα 1. Γεωγραφική αναπαράσταση της κοινής καταγωγής των πέντε από τις εννέα οικογένειες που φέρουν τη μετάλλαξη p.Cys618Arg. Το χωριό που βρίσκεται στο βορειοδυτικό άκρο της επαρχίας Λεμεσού αναφέρεται με κόκκινο χρώμα. Ο κοινός απλότυπος ο οποίος είναι συνδεδεμένος με την μετάλλαξη p.Cys618Arg καταδεικνύεται στο δεξιό κάτω μέρος της εικόνας.
Βιβλιογραφικές αναφορές:
- Skordis N, Neocleous V, Kyriakou A, Efstathiou E, Sertedaki A, Philibert P, Phylactou LA, Lumbroso S & Sultan C. The IVS1-2A>G mutation in the SRD5A2 gene predominates in Cypriot patients with 5alpha reductase deficiency. J Endocrinol Invest 2010 33 810-814.
- Neocleous V, Ioannou YS, Bartsota M, Costi C, Skordis N & Phylactou LA. Rare mutations in the CYP21A2 gene detected in congenital adrenal hyperplasia. Clin Biochem 2009 42 1363-1367.
- Fanis P, Skordis N, Frangos S, Christopoulos G, Spanou-Aristidou E, Andreou E, Manoli P, Mavrommatis M, Nicolaou S, Kleanthous M, Cariolou MA, Christophidou-Anastasiadou V, Tanteles GA, Phylactou LA & Neocleous V. Multiple endocrine neoplasia 2 in Cyprus: evidence for a founder effect. J Endocrinol Invest 2018.
Αρθρογραφία
Δρ. Παύλος Φάνης
Τμήμα Μοριακής Γενετικής, Λειτουργίας & Θεραπείας, Ινστιτούτο Νευρολογίας &
Γενετικής, Λευκωσία, Κύπρος
*Φωτογραφία από αριστερά: Δρ. Βάσος Νεοκλέους και Δρ. Παύλος Φάνης οι οποίοι συμμετείχαν στην μελέτη
This thread has been closed from taking new comments.